|
|
Atorvastatine Tahor° Parke Davis Pharmacia Searle Pfizer |
Cérivastatine+Cholstat° Fournier Staltor° Bayer |
FluvastatineFractal° Fabre Lescol° Novartis Sandoz |
PravastatineElisor° BMS Vasten° Aventis RPR Spécia |
SimvastatineLodalès° Sanofi Synthélabo Zocor° MSD |
|
Demi vie |
14 h |
2-3 h |
2.3 h +- 0.9 |
1 h 30 – 2 h |
2 h |
|
Liaison aux protéines plasmatiques |
95 % |
99.1 % - 99.5% |
>98 % |
50 % |
> 90 % |
|
Métabolites actifs |
Oui (70 % de l’activité inhibitrice) |
Oui |
Un métabolite hydroxylé avec activité pharmacologique mais que l’on ne retrouve pas dans le sang |
Non |
Oui, nombreux (dont bêta-hydroxyacide : 80 % de l’activité inhibitrice) |
|
Prodrogue |
Non |
Non |
Non |
Non |
Oui Lactone inactive hydroxylée dans le foie en forme hydroxyacide active |
|
Excrétion |
biliaire |
30% urinaire 70 % biliaire (fécès) |
6 % urinaire 70 % biliaire (fécès) |
20 % urinaire 70 % biliaire (fécès) |
biliaire |
|
|
AtorvastatineTAHOR° Parke Davis Pharmacia Searle Pfizer |
Cérivastatine+ CHOLSTAT° Fournier STALTOR° Bayer |
Fluvastatine FRACTAL° Fabre Sinbio LESCOL° Novartis |
Pravastatine ELISOR° BMS VASTEN° Aventis RPR Spécia |
Simvastatine LODALES° Sanofi Synthélabo ZOCOR° MSD |
|
DOSES |
20 – 40 mg |
0.1 0.3 mg |
20 – 40 mg |
20 mg |
5 – 20 mg |
|
POSOLOGIE |
20 – 80 mg |
0.1 – 0.3 mg |
20 – 80 mg |
10 à 40 mg |
5 à 40 mg |
|
CONTRE INDICATIONS |
Insuf. hépatique sévère Insuf. rénale sévère Grossesse |
Insuf. hépatique sévère Grossesse |
Insuf. hépatique sévère Insuf. rénale sévère Grossesse |
Insuf. hépatique sévère Insuf. rénale sévère Grossesse |
Insuf. hépatique sévère Grossesse |
|
PRECAUTIONS D’EMPLOI |
Tests hépatiques CPK |
Tests hépatiques CPK Adaptation poso, si insuffisance rénale sévère |
Tests hépatiques CPK |
Tests hépatiques CPK |
Tests hépatiques CPK Adaptation poso, si insuffisance rénale sévère |
|
EFFETS SECONDAIRES |
Musculaires : ↑CPK Myalgies++, faiblesse musc. Hépatiques : ↑transaminases++, gamma GT, phosphatases alcalines Digestifs : nausées, vomissements, douleurs abdom., diarrhées… Hypo-hyperglycémies Thrombopénies Impuissances, alopécies |
Musculaires : ↑CPK Hépatiques : ↑ transaminases+ |
Musculaires : ↑CPK Myalgies++, faiblesse musc. Hépatiques : ↑transaminases++, gammaGT, phosphatases alcalines Digestifs : nausées, vomissements, douleurs abdom., diarrhées |
Musculaires : ↑CPK Myalgies++, faiblesse musc. Hépatiques : ↑transaminases++, gammaGT, phosphatases alcalines Digestifs : nausées, vomissements, douleurs abdom., diarrhées… |
Musculaires : ↑CPK Myalgies++, faiblesse musc. Hépatiques :↑transaminases+++, gammaGT, phosphatases alcalines Digestifs : nausées, vomissements, douleurs abdom., diarrhées… Manifestations d’hypersensibilité : angiooedème, arthralgie, thrombocytopénie Paresthésies |
|
INTERACTIONS MEDICAMENT. |
Ciclosporine Fibrates Antivitamines K Digoxine Anti acides Contraceptifs Erythromycine Cholestyramine Inducteur cytochrome P450 |
Ciclosporine** Fibrates** **en cours d’étude |
Ciclosporine Fibrates Antivitamines K Cimétidine, ranitidine, oméprazole : ↑ biodisponibilité Rifampicine ↓biodisp. Inducteur cytochrome P450* |
Ciclosporine Fibrates Antivitamines K Inducteur cytochrome P450 |
Ciclosporine Fibrates Acide nicotinique Imidazolés Antivitamines K Warfarine Inducteur cytochrome P450 |
|
PRIX : CTJ |
4.06 à 16.28 FF |
|
4.06 à 16.28 FF |
3.73 à 14.92 FF |
3.73 à 14.92 FF |
*Les produits inducteurs du cytochrome P450 augmentent la dégradation hépatique d’autres molécules donc diminuent leur efficacité. Les produits inhibiteurs du cytochrome P450 diminuent la dégradation de certaines molécules donc augmentent leur toxicité
|
Rhabdomyolyse |
Principales
Causes :
|
| Complications
des rhabdomyolyses :
|
Welcome to my rhabdomyolysis page. This is actually a copy of an assignment I did for my fourth year medicine neuropathology course back in 1997. Whilst doing this assignment I found that there was not much information about this topic on the web, so I thought it might be useful to put online. So here it is, everything I was able to find about rhabdomyolysis. I hope you find it helpful. If you would like more information please speak to your doctor. I am not a specialist in this area and would not be of any help beyond what I have put in this web page. Nevertheless, I hope this information is of value.
OUTLINE
![]() LINKS
LINKS
![]() Dr
Baggas' Surgery
Dr
Baggas' Surgery
![]() Baggas'
World
Baggas'
World
INTRODUCTION
Rhabdomyolysis is a common disorder which may result from a large variety of
diseases, trauma, or toxic insults to skeletal muscle. It may be defined as a
clinical and biochemical syndrome resulting from an injury which damages the
integrity of the sarcolemma of skeletal muscle, leading to the release of
potentially toxic muscle cell components into the circulation.(1,2,3) This may
result in potential life-threatening complications including myoglobinuric acute
renal failure, hyperkalaemia and cardiac arrest, disseminated intravascular
coagulation, and more locally, compartment syndrome.
BIOCHEMISTRY
The primary diagnostic indicator of rhabdomyolysis is an elevated serum
creatine phosphokinase (CK) to at least five times the normal value.(2) This
elevation is generally to such a degree that myocardial infarction and other
causes of a raised CK are excluded. Additionally, the CK-MM isoenzyme
predominates in rhabdomyolysis, comprising at least 98% of the total value.(4)
The other important finding frequently seen in rhabdomyolysis is myoglobinuria.
Myoglobin, a haem protein which functions as an oxygen store in type 1 skeletal
muscle fibres, normally has a rapid renal clearance which maintains a low plasma
level up to a certain serum concentration.(5) As myoglobin is released into the
circulation from necrotic muscle cells it first becomes detectable in the urine
at serum concentrations ranging from 300ng/ml to 2 g/ml and produces visible
pigmenturia (classically a "coca-cola" coloured urine) at
concentrations exceeding 250 g/ml.(6) This discolouration is caused by myoglobin
plus metmyoglobin in the urine.(7) Biochemical tests for pigmenturia are
strongly suggestive of myoglobinuria in the absence of haemoglobinaemia and
haematuria.(7) Other important biochemical findings in rhabdomyolysis include
hyperkalemia, hypocalcaemia, hyperphosphataemia, hyperuricaemia, and raised
levels of other muscle enzymes including lactate dehydrogenase, aldolase,
aminotransferases, and carbonic anhydrase III (which is a very specific marker
for skeletal muscle injury).(2) Metabolic acidosis may result from release of
phosphate, sulphate, uric acid, and lactic acid from the muscle cell.(1)
AETIOLOGY
The causes of rhabdomyolysis can be broadly divided into hereditary (table
1) and acquired (table 2) groups. The hereditary
causes consist primarily of enzyme defects causing disorders of carbohydrate
metabolism(8), mitochondrial lipid metabolism(8), and other inherited disorders
such as malignant hyperthermia (8,9) and neuroleptic malignant syndrome(10).
Table 1 : Inherited causes of rhabdomyolysis. (from Poels and Gabreels (1993) Clin Neurol Neurosurg 95 : 175-192.
Deficiencies of glyco(geno)lytic enzymes
myophosphorylase (McArdle's disease)
phosphorylase kinase
phosphofructokinase (Tarui's disease)
phosphoglycerate mutase
phosphoglycerate kinase
lactate dehydrogenase
Abnormal Lipid Metabolism
carnitine palmitoyltranferase deficiency I and II
carnitine deficiency
Other genetic disorders
idiopathic rhabdomyolysis
myoadenylate deaminase deficiency
malignant hyperthermia
neuroleptic malignant syndrome
Acquired causes may be divided into traumatic, ischaemic, metabolic, infectious, inflammatory, and toxic groups(table 3) (11), as well as exercise and heat related causes.
Table 2 : Acquired causes of rhabdomyolysis. (from Poels and Gabreels (1993) Clin Neurol Neurosurg 95 : 175-192.
Toxic
alcohol
drugs and toxins (see Table 3)
Excessive muscle exercise
sports and military training
status epilepticus
status asthmaticus
convulsions
prolonged myoclonus, acute dystonia
Direct muscle injury
crush
burning, freezing
electric shock, lightning stroke
Ischemic injury
compression
vascular occlusion
sickle cell trait
Metabolic disorders
diabetic ketoacidosis
nonketotic hyperosmolar coma
hypothyroidism
hypophosphatemia
hyponatremia
hypokalemia
Infections
bacterial
viral
Heat-related syndromes
toxic shock syndrome
heat stroke
Inflammatory myopathies
polymyositis
dermatomyositis
Others
anticholinergic syndrome
withdrawal of L-Dopa
Table 3 :
Drugs and toxins known to cause rhabdomyolysis. (11)
Drug-induced coma, seizures,
dyskinesia Other drugs
Barbiturates, Amphetamines
Heroin, Phenmetrazine
Methadone, Phencyclidine
Glutethimide, Phenylpropanolamine
Chlorpromazine, Morphine
Diazepam, Dihydrocodeine
Rohypnol° LSD
Lithium, Salicylates
Amoxapine, Clofibrate / Bezafibrate
Phenelzine`, Epsilon-aminocaproic acid
Phenformin / fenfluramine, Isoniazid
Meprobamate, Loxapine
Antihistamines / paracetamol Theophyllin
Oxprenolol, Pentamidine
Ethanol, Vasopressin
Post-anaesthetic Toxins
Suxamethonium, Ethanol
Malignant hyperpyrexia, Isopropyl alcohol
Carbon monoxide
Neuroleptic malignant syndrome
Mercuric chloride
Haloperidol, Ethylene glycol
Stelaziine, Copper sulphate
Fluphenazine, Zinc phosphide
Other neuroleptics, Strychnine
Metaldehyde
Chloralose
Hypokalaemia
Paraphenylenediamine
Diuretics, Toluene (paint sniffing)
Carbenoxolone, Gasoline sniffing
Amphotericin B, Lindane / benzene
Liquorice, Snake bite
Hornet / wasp sting
Brown spider bite
Haff disease
Quail ingestion
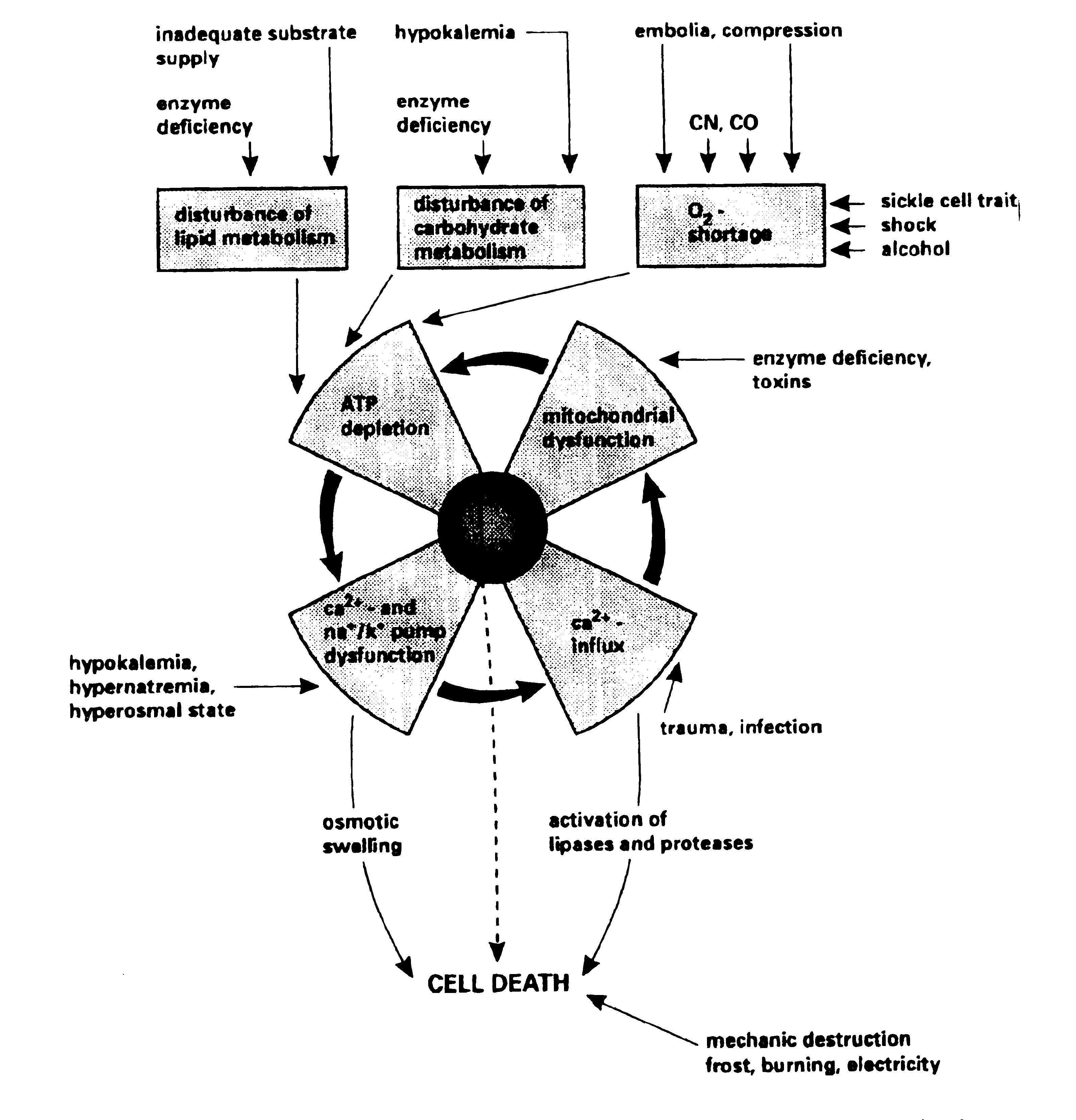
PATHOGENESIS
Although the causes of rhabdomyolysis are so diverse, the pathogenesis appears
to follow a final common pathway, ultimately leading to muscle necrosis and
release of muscle components into the circulation. Whatever the injurious
process, the end result is an increased cellular permeability to sodium ions due
to either plasma membrane disruption or reduced cellular energy (ATP)
production.(1) Accumulation of sodium in the cytoplasm leads to an increase in
intracellular calcium concentration (which is normally very low relative to the
extracellular concentration).(2) This accumulation of calcium is due both to
direct injury to the cell and to increased activity of an Na+/Ca2+ exchanger
protein which brings more calcium into the cell as it attempts to remove the
excess sodium. Depletion of ATP also contributes directly to calcium
accumulation due to a reduction in the activity of the Ca2+ ATPase which
normally acts to pump calcium out of the cell and sequester it in the
sarcoplasmic reticulum.(3)
Therefore, the common pathogenetic feature of all disease processes causing
rhabdomyolysis is an acute rise in the cytosolic and mitochondrial calcium
concentration in affected muscle cells, which sets off a chain of events that
ultimately results in muscle cell necrosis. This includes activation of
degradative enzymes such as phospholipase A2 (PLA) and neutral proteases,
leading to membrane phospholipid and myofibril damage.(3) Jackson et al. (12)
suggest that the most significant of these is activation of PLA, and that most
of the membrane and mitochondrial damage in rhabdomyolysis can be attributed to
this. PLA mediated attack on mitochondrial and sarcolemmal membrane
phospholipids leads to the formation of lysophospholipids and free fatty acids.
These further potentiate the injury by causing direct membrane damage themselves
and through alterations in ionic transport, which results in further influx of
sodium and calcium.(3) Thus the reaction becomes self-perpetuating. Depletion of
ATP and mitochondrial damage may be the primary event which sets off this
cascade (as in most hereditary causes of rhabdomyolysis and exertional
rhabdomyolysis) or it may occur secondarily to the rise in calcium
concentration. Either way, mitochondrial damage and depletion of ATP contributes
to the pathogenesis via the following :
(1) Failure of Ca2+ ATPase leading to failure of calcium sequestration and
reduced efflux of calcium from the cell.
(2) Failure of Na+/K+ ATPase leading to increased intracellular sodium and
increased Na+-Ca2+ exchange, further contributing to the increased intracellular
calcium.2
(3) Generation of toxic oxygen free radicals such as superoxide causes further
cellular damage.(3)
A simple schematic representation of these processes is shown in Figure
1 . Ultimately, the combination of all of these processes is a
self-sustaining reaction which results in muscle cell lysis (figure2)
and release of intracellular components into the extracellular fluid and
circulation.(3) Locally, accumulation of these products may result in
microvascular damage, capillary leak and increased intracompartmental pressures,
and reduced tissue perfusion and ischaemia, which may further potentiate the
muscle damage.
As has already been stated, there are many different causes of rhabdomyolysis,
and although the final reaction is fairly stereotyped, the mechanism by which
this reaction is triggered is quite variable. I will now discuss some of the
specific hereditary and acquired causes of rhabdomyolysis in more detail.
HEREDITARY CAUSES OF RHABDOMYOLYSIS
Disorders of Muscle Carbohydrate Metabolism
The first genetic disease described which causes rhabdomyolysis is McArdle's
disease (myophosphorylase deficiency), an autosomal recessive condition in which
there is selective necrosis of type 2 muscle fibres.(8) These fibres are more
dependent on glyocolysis for generation of ATP and therefore will be more
sensitive to an enzyme defect which prevents the formation of glucose from
glycogen. Hence it is ATP depletion which is responsible for rhabdomyolysis in
this disease. Muscle pain and rhabdomyolysis are induced by vigorous
exercise,and relieved by rest in this disease, consequently patients can adjust
their life styles to prevent symptoms by avoiding vigorous exercise which
requires activation of type 2 fibres. Other inherited diseases affecting the
glycolytic/ glycogenolytic pathways include
phosphofructokinase deficiency (Tarui's disease), and phosphoglycerate mutase
deficiency.(8)
Carnitine Palmitoyltransferase Deficiency
Where the disorders of carbohydrate metabolism affect primarily anaerobic type 2
muscle fibres, diseases of lipid metabolism such as Carnitine
palmitoyltransferase deficiency (CPD), have a greater effect on aerobic
type 1 fibres which depend on the oxidation of long chain fatty acids to produce
energy. CPD, an autosomal recessive disorder, has been shown to be the most
common hereditary disease causing rhabdomyolysis.(3) In this disease muscle pain
and rhabdomyolysis develop after prolonged exercise with inadequate nutrient
intake, not in the initial phase as in the glycogen storage disorders. Treatment
of this disease involves frequent high carbohydrate meals and avoidance of
prolonged exercise.(8)
Malignant Hyperthermia
Another genetic disease which may result in rhabdomyolysis is malignant
hyperthermia (MH) (Figure
3). In this disease, episodes of hyperthermia and rhabdomyolysis are
triggered by exposure to volatile anaesthetics such as halothane, or
succinylcholine, a depolarising muscle relaxant.(9) MH appears to be an
autosomal dominant condition with variable penetrance(7), and may involve a
defect in the ryanodine receptor of the calcium release channel of the
sarcoplasmic reticulum.(7) These patients have higher than normal resting
sarcoplasmic calcium concentrations, and exposure to the above agents may
trigger further uncontrolled calcium release, leading to excessive muscle
contraction, hyperthermia, and rhabdomyolysis.(8) The diagnosis of MH
susceptibility can be made only by muscle biopsy and a positive in vitro
response to provocative agents such as halothane, succinylcholine, and caffeine.
This in vitro response shows a patchy, moth-eaten appearance of type 1
fibres(13) (Figure 4).
Type 1 fibres are predominantly affected in MH due to their lower capacity for
anaerobic metabolism, and therefore more rapid ATP depletion in the
hypermetabolic state of MH.
Neuroleptic Malignant Syndrome
A similar disorder is the Neuroleptic Malignant Syndrome (NMS), in which there
is a gradual development of hyperthermia, muscle rigidity, fluctuating
consciousness, and autonomic instability.(10) Rhabdomyolysis and myoglobinuria
may result. Drugs which can cause NMS include phenothiazines, butyrophenones,
and other antipsychotics and antidepressants. It is believed that the underlying
defect in NMS may be a central or presynaptic one, in contrast to the peripheral
defect in MH. (10)
ACQUIRED CAUSES OF RHABDOMYOLYSIS
There are also many non-hereditary causes of rhabdomyolysis, which are much more
common than the hereditary causes.
Exertional Rhabdomyolysis
Exertional rhabdomyolysis and heat stroke are probably the most common causes of
severe rhabdomyolysis. This occurs most commonly in untrained people undertaking
vigorous exercise in hot, humid weather.(3) The pathogenesis of rhabdomyolysis
in these cases appears to be due to a combination of mechanical and thermal
muscle injury and ATP depletion, both of which ultimately lead to calcium
accumulation. Excess muscle activity may also lead to rhabdomyolysis in
conditions such as generalised seizures, status epilepticus, status asthmaticus,
myoclonus, and severe dystonia. (2)
Crush Injury and Trauma
In crush injury and other forms of trauma, rhabdomyolysis is generally due to
direct muscle injury and ischaemia. However, in addition to this, in the crush
injury, reperfusion after prolonged ischaemia is also believed to play a
significant role in muscle damage.(14) This is believed to be mediated by the
formation of oxygen free radicals, the action of granulocytes, and increased
calcium uptake after ischaemia (which is due to exchange of calcium for excess
intracellular sodium which has accumulated during the ischaemic period).
Alcoholism
Alcoholism is another common cause of rhabdomyolysis. This may be secondary to
to alcohol related trauma, seizures, or coma, or may be due to a direct toxic
effect of ethanol on skeletal muscle, resulting in both a chronic myopathy, and
acute rhabdomyolysis.(3)It is believed that ethanol causes direct sarcolemmal
injury, leading to increased sodium permeability, and subsequent accumulation of
calcium.1 Hypophosphataemia may be an important precipitant of rhabdomyolysis in
alcoholics, since the ability of muscle cells to produce ATP would be reduced.
(4)
Drugs and Toxins
A large range of drugs and toxins have been seen to cause rhabdomyolysis. Many
of these are listed in Table 3. The mechanisms of muscle damage in these
instances are diverse.
Some drugs appear to have a direct toxic action on skeletal muscle when given
systemically. These include cholesterol lowering drugs (clofibrate, gemfibrozil,
HMG CoA reductase inhibitors), emetine (ipecac), zidovudine (AZT), vincristine,
and epsilon-aminocaproic acid (Figure
5).(15,11)
An immunological mechanism may be responsible for the myositis seen in patients
treated seen in patients treated with D-penicillamine, L-tryptophan, and rarely
in other drugs including procainamide, cimetidine, phenytoin, and levodopa.(11)
Amphotericin B, carbenolexone, liquorice, laxatives, and diuretics may cause
rhabdomyolysis secondary to sever hypokalaemia.(11)
Another mechanism by which drugs may cause rhabdomyolysis is by excessive
neuromuscular stimulation. These drugs include phencyclidine (PCP), and
acetylcholinesterase inhibitors.(11)
Drugs such as heroin and barbiturates may contribute to rhabdomyolysis via coma
and muscle compression following overdose.(2)
In addition to the range of pharmacologic agents which cause rhabdomyolysis, it
can also be caused by the venoms of a number of snakes, spiders, and wasps.(11)
Microbial toxins such as the a-toxin of Clostridium perfringens (gas gangrene),
can also cause rhabdomyolysis, as can excessive consumption of quail. (11)
CLINICAL FEATURES
The clinical features of rhabdomyolysis are quite variable, no doubt due to the
large range of causes of this condition. Broadly, they can be divided into the
following2 :
(1) Muscular signs and symptoms
(2) General internal disturbances
(3) Complications
Muscular signs and symptoms
These include pain, weakness, tenderness, and contractures. This may involve
specific groups of muscles or may be generalised. Most frequently the involved
muscle groups are the calves and lower back, however a significant proportion
may show no signs of muscle injury at all.(16) Sometimes haemorrhagic
discolouration of the overlying skin may be seen. Typically the muscle disorder
is self-limiting and resolves within days to weeks, due to the regenerative
capacity of muscle.
General internal disturbances
These include malaise, fever, tachycardia, nausea, and vomiting. Hyperuricaemia
may lead to encephalopathy with depression of respiration with hypoxia and
respiratory acidosis. (2)
COMPLICATIONS
The complications of rhabdomyolysis are due to the local effects of muscle
injury, and the systemic effects of released muscle components. These include :
(1) Hypovolaemia - due to haemorrhage, and influx of fluid into necrotic
muscle. 4-11 litres of normal saline may be required to maintain cardiac and
urine output. (2,16)
(2) Cardiac arrest and arrhythmias - Hyperkalaemia can precipitate severe
arrhythmias and cardiac arrest. This toxicity is potentiated by the
hypocalcaemia resulting from calcium deposition in necrotic muscle. Therapy
often involves the use of ion exchange resins.
(3) Compartment Syndrome - (Figure
6)in acute rhabdomyolysis muscle swelling within a tight fascial compartment
can lead to compression of vessels and nerves. This can lead to nerve damage and
muscle ischaemia due to reduced capillary flow. Ischaemia will result in further
oedema which prolongs the cycle. Prolonged ischaemia and infarction of muscle
tissue can result in replacement of muscle by inelastic fibrous tissue and
severe contractures (Volkmann's contracture).(17,2) The treatment of suspected
compartment syndrome is urgent decompression by open fasciotomy.
(4) Disseminated intravascular coagulation - this is an almost universal
finding in patients with rhabdomyolysis (18) and is probably due to activation
of the clotting cascade by released muscle components. Fortunately, in most
cases, the diagnosis of DIC is made purely by laboratory abnormalities rather
than overt clinical bleeding or thrombosis.(16)
(5) Acute Renal Failure - this is probably the most
significant and most feared complication of rhabdomyolysis, and is said to occur
in about 30% of patients.(16) Conversely, rhabdomyolysis has been said to be a
factor in 8% of cases of acute renal failure2 so this is by no means an uncommon
condition. The mechanisms of myoglobinuric acute renal failure have been
comprehensively explored by Zager (1996) (3) and include the following :
(1) Renal vasoconstriction/hypoperfusion - due to hypovolaemia and haem-
protein induce renal tubular ATP depletion
(2) Haem protein cast formation - precipitation of pigment casts in
distal tubules may contribute to acute tubular necrosis, especially in aciduria
(3) Ischaemic tubular injury - independent of haemodynamic influences,
haem protein can potentiate proximal tubular ischaemic damage
(4) Haem iron induced oxidant stress - intratubular release of haem iron
catalyses formation of toxic oxygen free radicals
Prevention of myoglobinuric ARF involves maintenance of circulating blood volume
by adequate fluid replacement of up to 11 litres of normal saline. (2)
Administration of frusemide and/or mannitol is used to maintain a diuresis and
enhance haem protein elimination. Alkalinization of the urine by the addition of
sodium bicarbonate to the intravenous fluids has been suggested (since acidic
urine favours myoglobin nephrotoxicity) however this is controversial since
bicarbonate may aggravate existing hypocalcaemia. (2,3)
CONCLUSIONS
Rhabdomyolysis is a common condition which complicates a a variety of genetic
and acquired diseases. It is characterised by muscle cell necrosis and release
of muscle cell components into the circulation, most notably creatine
phosphokinase (CK) and myoglobin. The primary mechanism through which muscle
damage occurs in rhabdomyolysis is sarcoplasmic calcium overload leading to
activation of degradative enzymes. This may occur secondary to a number of
processes including ATP depletion and increased intracellular sodium
concentration, and direct sarcolemmal injury. The complications of
rhabdomyolysis can be potentially life threatening, and include cardiac arrest
and myoglobinuric acute renal failure. Prompt action must be taken to prevent
these complications in a patient with rhabdomyolysis, most importantly
aggressive intravenous volume replacement.
REFERENCES
1. Anderson, J.R. (1985) Atlas of Skeletal Muscle Pathology. Lancaster : MTP Press.
2. Apley, A.G. and Solomon, L. (1994) Concise System of Orthopaedics and Fractures. Oxford : Butterworth-Heinemann.
3. Argov, Z. and Mastaglia, F.L. (1994) Drug-induced neuromuscular disorders in man. In: Walton, J., Karpati, G., and Hilton-Jones, D. (Eds) Disorders of Voluntary Muscle (6th ed). New York : Churchill Livingstone, 989-1029.
4. Baxter, L.R.and Guzé, B.H. (1985) Neuroleptic Malignant Syndrome. NEJM 313(3): 163-166.
5. Brumback, R.A., Feeback, D.L., and Leech, R.W. (1992) Rhabdomyolysis in childhood. Paediatric Neurology 39(4) : 821-858.
6. Dayer-Berenson, L. (1994) Rhabdomyolysis : A comprehensive guide. ANNA Journal 21(1): 15-18.
7. Edwards, R.H.T., Jackson, M.J. and Jones, D.A.(1984) Experimental skeletal muscle damage : the nature of the calcium activated degenerative processes. Eur J Clin Invest 14: 369-374.
8. Gabreëls, F.J.M. and Poels, P.J.E (1993) Rhabdomyolysis : a review of the literature. Clin Neurol & Neurosurg 95: 175-192.
9. Gronert, G.A. (1986) Malignant Hyperthermia. In: Engel, A.G, and Banker, B.Q. (Eds) Myology, Vol 2. New York : McGraw-Hill, 1763-1784.
10. Kakulas, B.A. and Mastaglia, F.L. (1992) Drug-induced, toxic and nutritional myopathies. In: Mastaglia, F.L. and Walton, J. (Eds) Skeletal Muscle Pathology (2nd ed). New York : Churchill Livingstone, 511-540.
11. Knochel, J.P. (1990) Catastrophic medical events with exhaustive exercise : "White collar rhabdomyolysis". Kidney International 38: 709-719.
12. Knochel, J.P. (1992) Hypophosphataemia and rhabdomyolysis. JAMA 92: 455-457.
13. Knochel, J.P. (1993) Mechanisms of rhabdomyolysis. Current Opinion in Rheumatology 5: 725-731.
14. Moxley, R.T. (1994) Metabolic and endocrine myopathies. In: Walton, J., Karpati, G., and Hilton-Jones, D. (Eds) Disorders of Voluntary Muscle (6th ed). New York : Churchill Livingstone, 647-716.
15. Odeh, M. (1991) The role of reperfusion-induced injury in the pathogenesis of the crush syndrome. NEJM 324(20) : 1417-1422.
16. Penn, A.S. (1986) Myoglobinuria. In: Engel, A.G, and Banker, B.Q. (Eds) Myology, Vol 2. New York : McGraw-Hill, 1785-1805.
17. Saad, E.B. (1997) Rhabomyolysis and Myoglobinuria. (internet reference : http://www.medstudents.com.br/terin/terin3.htm)
18. Zager, R.A. (1996) Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney International 49 : 314-326.
NOTE : To any fourth year UWA medical students contemplating plagiarising this page to use as your assignment, please think again. I put this page up to make up for the lack of information about rhabdomyolysis on the Web, and a lot of people have benefitted from it. Feel free to use this information as part of your own research, just don't copy it verbatim to save yourself getting in trouble. Thanks.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}